


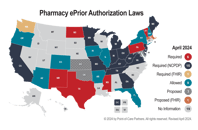
As state legislatures intensify efforts to reform prior authorization processes, these changes may reflect broader regulatory trends such as those outlined by CMS in rules like...

REMS Automation: Advancing Patient Safety Through Collaboration
Patient safety is always the top priority, and there are many programs in place to support continuous improvement. As a consultant with POCP and subject matter expert on the CodeX...

Another Year on the NCPDP Board of Trustees: Reflecting & Looking Ahead
As I reflect on another impactful year on the National Council for Prescription Drug Programs (NCPDP) Board of Trustees, the connection between my role at Point-of-Care Partners...

Reflecting on HIMSS24: Insights and Innovations from the Front Lines
As spring blossoms each year, the healthcare IT community gears up for one of its most anticipated events: the Healthcare Information and Management Systems Society (HIMSS)...

From Federal to State: The Vital Importance of Vigilance in DEA Rule Changes and Policy Shifts for Telehealth Substance Use Disorder Treatment
The Controlled Substances Act (CSA) in the United States imposes stringent regulations governing the prescribing, dispensing, and usage of narcotic drugs, especially those...

POCP at ViVE24: Sessions and Perspectives
As the countdown to ViVE24 begins, the healthcare industry is gearing up for a deep dive into the latest trends and innovations shaping the future of patient care. Point-of-Care...

HHS Data Strategy: The Vital Role of Consent Management in Bridging Health & Human Services Data
The latest HHS Data Strategy was unveiled back in December, and it's packed with updates that provide valuable insight into their priorities and where they're putting their...

CMS Rule Drives New Era of API-Enabled Data Sharing & Prior Authorization Reform
On January 17, 2024, the Centers for Medicare & Medicaid Services (CMS) unveiled a long-awaited final rule aimed at advancing interoperability and enhancing prior authorization...
Washington State Takes a Leap to Require FHIR APIs and Reshape Prior Authorization
Washington State has taken a pioneering leap in reshaping the landscape of prior authorization, racing ahead of federal guidelines by mandating the use of FHIR (Fast Healthcare...

First-Hand Reflections on the HITAC Pharmacy Interoperability & Emerging Therapeutics Task Force
Last month, we concluded our work on the HITAC Pharmacy Interoperability & Emerging Therapeutics Task Force. In June, the task force embarked on a mission fueled by the...